众所周知,药物进入体内后,可以在各种酶的催化下发生代谢转化,产生结构相似的代谢产物。虽然结构与母药相似,但代谢物肯定是项目研发中的新化学成分。通过动物和体外方法对候选化合物进行毒理学研究,通常更关注母体药物的特性,而不是代谢产物。而且动物物种和人体可能存在差异,不能简单地假设产生的代谢产物和人体产生的是一样的。当人体内某些代谢产物的暴露量与动物种类不成比例时,就会存在安全风险。因此,提出了安全性检测中代谢物的概念。
自20世纪90年代初以来,液相色谱-质谱联用技术(LC-MS)取得了突破性的发展,创新了药物代谢领域的技术手段,使得在血浆等复杂底物中鉴定代谢物变得相对容易。再加上放射性标记的ADME(吸收、分布、代谢和排泄)研究,可以生成血浆、尿液和粪便的定量代谢物谱,使得代谢物研究成为药物研发的重要组成部分。
虽然开发团队都知道一般药物的代谢物可能具有药理活性或毒性作用,需要进行研究,但具体到项目,它们面临以下问题:
1.哪些代谢物需要纳入安全性评价范围?
2.是否有代谢物暴露的阈值来确定进一步研究的必要性?
3.有必要获取代谢物的化学结构吗?
这些都需要有相关的法律法规或者指导原则来遵循。
MIST指导原则的变迁
2002年,备受人体代谢物安全性困扰的行业率先“出击”。来自不同公司和组织的一组科学家联合发表了一份关于药物代谢物风险评估的提案(参见第2条)。文章提出,在临床前毒理学物种研究中,必须涵盖人体血浆中暴露量占药物相关物质总量25%以上的代谢物。这是关于薄雾的第一次论述。
2008年,FDA终于发布了MIST的指导原则,将人体血浆中代谢物的阈值调整为药物原型的10%。
2010年,ICH在综合了更广泛的意见后发布了指导意见,提出在进行大规模临床试验前,有必要对人体血浆中暴露量占药物相关物质总量10%以上的代谢物进行非临床安全性研究(ICH M3(R2))。FDA随后跟进并于2016年修订了指南,以符合ICH。

毫无疑问,这些指导原则可以促进药物安全性研究,但更重要的是,它们为药物研发提供了一个研究代谢产物的框架。从临床前到临床,每个阶段要解决的药物代谢问题是不一样的,可能会因策略和靶点的不同而不同。比如肿瘤晚期、耐药HIV(艾滋病病毒)、先天性酶缺乏症等严重疾病的药物研发等。对代谢物的安全性研究有相应的简化措施。对于常规项目,代谢物的典型策略可分为三个阶段:
第一阶段(前IND)
现阶段药物代谢研究主要集中在体外和体内动物,如肝微粒体、肝细胞、肝S9、重组酶或血浆。将这些体外模型用于孵育代谢物,结合保留时间和质谱裂解规律,比较不同物种与人类之间代谢物的定性/定量差异。在此基础上,选择合适的物种进行毒理学研究。或者研究给药后动物的代谢产物,补充体外研究。动物的体内结果不能直接转化为准确的临床预测,但通过分析动物的主要代谢产物,我们可以判断代谢产物谱是否能覆盖人类的结果,这对临床安全性研究非常重要。
第二阶段(临床阶段I)
来自临床I期多剂量递增试验(MAD)的稳态血浆样本可用于人体代谢物的研究,这通常是首次获得药物的人体代谢物数据。虽然LC-MS的灵敏度可以满足痕量代谢物的分析,但是合成代谢物标准品的工作量太大,而且代谢物的相对丰度
如何判断《指导原则》中的代谢物是否超过药物有关物质总量的10%?如果将母药和所有代谢物定量相加,需要大量的合成标准品,这显然是不现实的。一般来说,在这个阶段,不需要精确量化。只需将NOAEL(无明显有害剂量)下的毒性种代谢物结果与临床疗效剂量下的血浆代谢物结果进行比较,即可确定是否存在临床前未覆盖的代谢物。常见的方法有两种,一种是用汉密尔顿池法计算代谢物的“暴露倍数”;另一种是依靠同位素标记试验,根据啮齿动物同位素标记的物质平衡结果,与大鼠同位素标记代谢物的浓度进行比较,得到因子,再换算成人代谢物的浓度。
一旦确定了临床前代谢物的覆盖范围,就可以决定是否需要合成代谢物,是否需要进行专门的体内和毒理学研究。但很多研究团队选择将这部分工作放回去,在完成人体同位素ADME的研究后再做决定,甚至将代谢物的研究放在临床概念验证(PoC)之后,也算是避免了因PoC的失败而做过多的“无用功”。
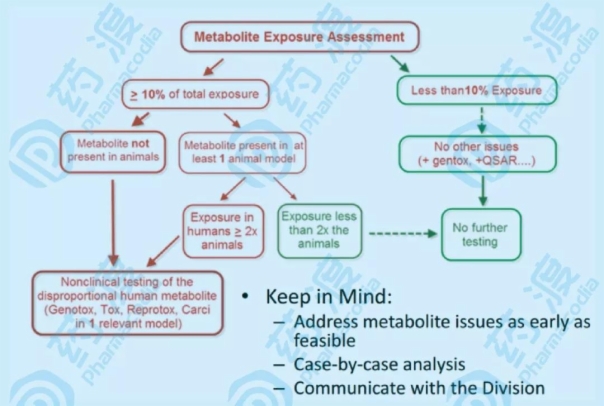
第三阶段(临床阶段II之后,临床阶段III之前)
因为主要代谢物的研究必须在临床扩大前完成,所以所有必要的代谢物研究都应该在这个阶段完成。包括代谢物的量化、通过高分辨率质谱测定未知代谢物、研究人类同位素标记ADME(其也可用于确定哪些代谢物需要DDI评估)以获得所有代谢物的光谱等。
对MIST的一些思考
自2008年FDA发布MIST指南至今已有十余年。在这十年间,代谢物和代谢安全的重要性逐渐深入人心。制药行业已经适应了MIST指南的要求,大量的案例也为R&D团队的代谢物研究计划提供了很好的参考。然而,遇到了许多问题。首先,很少有项目真正对代谢物进行毒理学研究,也很难找到循环代谢物直接导致安全问题的案例。很多情况下,明明安全性问题已经指向了代谢物,但最后却没有完整的证据。一方面,这可能是由于难以区分代谢物与母体药物的性质;另一方面,对于一些活性中间代谢物,常规分析方法有时无法检测出来,造成很大困难。大鼠服用恩格列吡嗪后,观察到明显的代谢产物介导的毒性,但未发现活性代谢产物和下游代谢产物。
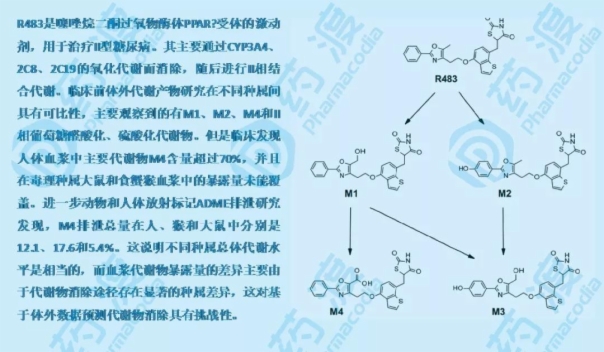
其次,反应性代谢产物或下游产物(如谷胱甘肽或半胱氨酸结合物的代谢产物)由于分子量大、亲水性强,通常通过胆汁和/或尿液快速排泄,因此很难根据MIST的要求比较不同物种间反应性代谢产物的暴露差异。此外,在一些特定项目中,目标组织中代谢物的测定可能更重要,但在人体中很难进行。换句话说,随着技术的发展,对代谢物的研究应该做到什么程度?MIST所做的一切是否真的影响了药物安全性的评价,是我们今后需要思考和解决的问题。

指导原则只是提供了一个方向。对于每一个项目,都需要设计一个具体情况具体分析的研究方案。除了常规毒性外,我们还应注意代谢物潜在的DDI发生。吉非罗齐的葡萄糖醛酸化代谢产物抑制西立伐他汀的代谢清除(主要由CYP2C8和OATP1B1介导),导致严重后果。此外,还有丁苯丙酮的代谢产物对CYP2D6表达水平的影响,以及去甲基地尔硫卓对CYP3A4的抑制作用。FDA和EMA的指南建议,当代谢物的体内水平达到母体药物的25%(FDA,2012)或暴露量超过药物总暴露量的10%(EMA,2012)时,需要研究代谢物的CYP抑制和转运体抑制。在2017年FDA新指导原则草案中,进一步要求对警示化学结构(官能团)进行TDI(时间依赖性抑制)研究;对于极性小于母体药物且暴露量大于母体药物25%的代谢物,以及极性大于母体药物且暴露量大于母体药物的代谢物,需要进行体外DDI的研究。
总之,MIST指导原则的引入不仅是DMPK科学家的研究参考,而且在很大程度上也是药学和药理学科学家对代谢物安全性的足够重视。只有这样,新药的安全性评价才能更加彻底,这也是MIST对药物研发的最大贡献。
参考文章:
[1]迷雾中的十年:从“安全性试验中的代谢物”监管指导下的药物开发中的药物代谢物研究中获得的经验。DOI:
[2]安全性试验中的药物代谢物。DOI:10.1006/
[3]安全性试验中的代谢物。
[4]FDA-工业用药物代谢物安全性测试指南。
[5]M3工业部(R2)关于进行人体临床试验和药品上市许可的非临床安全性研究的指南。
[6]安全性试验中的代谢物:临床药理学家的“迷雾”。doi:10.1038/