QbD理念的诞生
QbD,即品质源于设计(Quality by Design),起源于20世纪70年代丰田公司为提高汽车品质而提出的创造性概念,通过在通信、航空等领域的发展逐渐形成。对于制药业来说,这一概念最早出现在ICH Q8中:“一种系统的开发方法,始于预先确定的目标、产品和过程的理解和进行。”Ess控制基于完善的科学和质量风险管理,是基于充分的科学知识和质量风险管理的系统化研发方法。从预定的目标出发,强调对产品和过程的理解和过程控制”。2005年,FDA开始积极倡导和推广FDA。
QbT理念与QbD理念
QbT(质量源于检测)
QB代表“通过测试获得质量”,即质量来自于测试。认为质量是通过测试注入到产品中的,产品的质量取决于最终的测试。其基本控制框架见下图1。
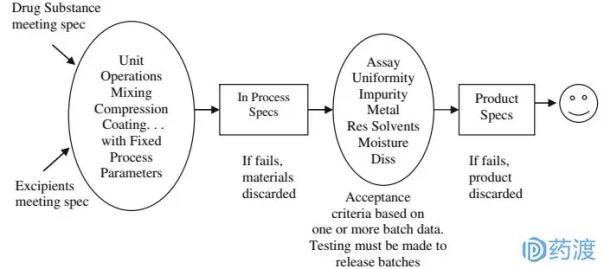
图1:1:QbT系统控制质量框架图
在该系统中,产品质量保证主要通过以下步骤完成:
原材料检测;
活性成分的生产;
固定制剂生产技术;
中间体(过程中的物质)的检测;
最终产品检验。
其中,起始原料的检测主要包括原料(有效成分)和辅料的检测。一般来说,这些原料只有满足生产商、FDA或其他标准(如USP)的要求才能被采用,但一般不会被进一步认可。因此,厂商为了降低风险,一般会制定更严格的内控标准,但严格标准的具体来源自己可能并不清楚。所以如果原料稍微晚一点变化,就可能导致制剂厂家。
成品的质量保证也是通过检测和评估来确定的,看检测结果是否符合厂家和FDA的标准要求。如果没有,药品就报废了;而失败的根本原因也一直没有完全搞清楚。出现一批故障后,厂家在不了解故障原因的情况下,仍然冒险继续生产,直到找到故障根源并解决;或者FDA批准修订相关可接受标准的补充申请(如放宽部分标准限值),同时对之前“不合格”的批次进行重新合格。
同时,为了保证制剂中间体符合FDA批准的相关标准,生产企业要对大量中间体进行检测(如混合均匀度BU、片剂硬度)。制造商不得随意更改批记录中的生产参数,也不得更改任何生产工艺,除非已向FDA发起变更补充申请并获得FDA批准;由于这个原因,FDA面临着大量的CMC补充申请变更。例如,从2005年到2006年,FDA仿制药办公室收到了3000多份CMC补充申请。
在QbT概念下,许多参数是固定的。固定的(或不灵活的)生产工艺和大量的测试样品是保证产品质量的关键手段,同时也要求生产工艺的一致性。总之,由于整个R&D方案和生产方案的认知度不高,出现了一套硬性的、不灵活的可接受标准,很多产品本身符合临床要求,却因为这个硬性的质量标准而被禁止放行。或者厂家选择向FDA发起补充申请,修改相关标准。另一方面,生产厂家对其产品所用的原辅材料了解不多,对生产工艺也不太了解,无法识别哪些性能或参数或步骤是影响产品质量的关键因素。但是这些关键信息无法正确传递给FDA的审评人员,所以FDA的审评人员只能采取保守的方式。
鉴于这种监管模式,所有的产品质量保证似乎都只与检测标准有关,而没有考虑到消费者的风险;这种监管模式导致生产厂家和审查人员在低风险产品上花费了过多的时间和资源,而对于风险相对较高的产品却没有足够的时间和资源。可以说,这种监管模式的结果是所有产品的资源平均分配,而实际上有些产品可能不需要那么多资源,而有些产品则需要更多资源。比如,对于审评者来说,他们认为对于剂型相对复杂的产品(如控释制剂、透皮制剂)或治疗窗较窄的产品,与其他剂型(如口服固体速释制剂)没有明显区别。事实上,剂型复杂的产品或者治疗窗窄的产品,在某些方面可能需要更深入的研究,而普通产品可能不需要花太多时间去研究影响不大的地方。
综上所述,传统的产品质量保证模式归因于生产过程的不灵活和对产品检测和控制的依赖;但是,在保证产品质量的前提下,如何提高产品的有效性和产品生产的效率,却很少或根本没有得到重视。因此,对于工艺或剂型复杂的产品,无法很好地识别放大过程中的关键点。产品标准也是通过一批或多批数据检测建立的。
QbD(质量源于设计)
ICH Q8指出,产品的质量不能由检验给出,而是由设计给出。了解这一点非常重要。ICH Q6A强调质量标准“规范”的作用:质量标准是一项重要的质量指示,由制造商提出并论证,经管理机构批准,作为批准产品的依据。所以,起决定作用的是生产厂家,而不是FDA批准。
QbD是一种基于风险的系统、科学、全面、主动的药物开发方法。从产品概念到产业化都是精心设计的,是对产品属性、生产工艺、产品性能之间关系的透彻理解。这意味着从药物研发的开始阶段就要考虑最终产品的质量,包括处方设计和开发、生产工艺路线的选择和确定等。QbD是从患者的角度识别产品的关键质量属性,并将其转化为药物研发和生产的控制因素,主要包括以下步骤:
确定目标药物质量概况的QTPP
设计开发处方和生产工艺,并系统评估、了解和改进。
识别关键质量属性CQAs、过程参数和变量来源。
结合风险管理制定适当的控制策略。
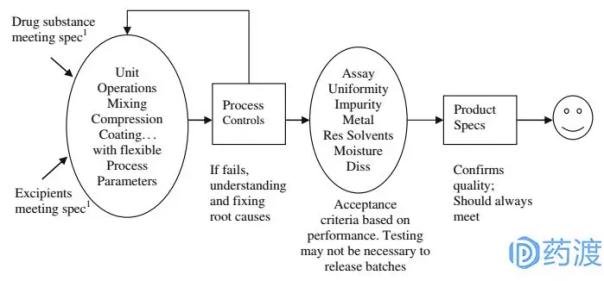
图2: QBD框架图
图2是QbD的框架图。与QbR不同,QbD认为产品质量标准的建立应结合临床相关性和产品性能,对生产工艺和产品质量标准进行提前设计和控制。药品质量的保证是一个系统的研究方法,依赖于对处方工艺的理解和控制,而最终产品检验只是保证产品质量的一致性,而不是生产工艺或过程控制的一致性。因此,基于QbD的标准总体上是合理的、严格可控的。
目前我们可以接受的内容设置标准和QbD的思路很像。一般我们通常把产品含量的可接受标准定在90%-110%,个别产品因为临床原因定的标准是95%-105%。这个标准不会因为实际测试数据而改变:即使大部分测试数据都集中在98%-100%,我们还是会把含量的可接受标准定在98%。然而,另一方面,我们似乎根据QbT概念设定了可接受的解散标准。在溶出度标准的制定过程中,我们主要基于实验室批次数据或小批量数据,试图找到具有分辨力的溶出曲线,而忽略(或不太关注)溶出曲线与药物体内行为的相关性。根据QbD的概念,溶出曲线的建立应尽可能反映药物在体内的行为。对于BCS和口服固体速释制剂,体内吸收限制步骤取决于药物的吸收过程,而具有快速溶出特性的药物不是体内吸收限制步骤。因此,他们的验收标准可以比实际测试数据更宽。同样,对于BCS II和IV药物,需要仔细选择尽可能反映内部环境的溶出条件和可接受的标准。
杂质(或降解产物)的可接受标准是药物的另一个CQAs,安全性是其标准建立的底线。根据QbD概念,杂质的可接受标准应该是基于其定量极限或生物安全级别(四个级别,其中I级危害最小),而不是基于实际检测得到的实验记录或批次记录。一些杂质的可接受标准也可以通过参考RLD制剂来建立(参考文章ANDA:根据FDA考虑杂质的要求)。
据悉,为了申报海外注册,上海医药集团新一医药公司已经在两个项目中使用了QbD。资料显示,传统方法无法稳定控制项目质量,一次通过率低。采用QbD后,50批次产品一次合格率100%。因此,从长远来看,实施QbD可以有效提高药品质量的合格率和稳定性,从而降低生产成本。所以,QbD的理想状态是一个双赢的结果:对于生产者来说,可以减少监管压力,降低生产成本;对于监管者来说,可以在不牺牲质量的情况下减轻监管压力;对于患者来说,可以获得有效的药物,更好的保证产品质量。
小结
在药物研发和建立质量标准的过程中,QbT和QbD遵循完全不同的理念:QbT理念下的产品质量标准是基于一系列小批量数据。如果这些数据是可接受的,则应为其制定相应的可接受标准,后续每个生产批次的检测结果都应符合这些标准,从而通过合格的检测结果来保证最终产品质量与生产工艺的一致性;在QB理念下建立的质量标准并不要求每一批都要检测,因为它的标准是建立在对处方工艺的理解或工艺控制的充分证明的基础上,更加灵活。
QbD的实施需要企业对生产流程进行深入的研究,同时也需要在硬件上进行一定的投入。但目前国内的企业都在“赶审批”,没有时间、精力和资金进行如此细致深入的研究。看起来他们没有QbD也能活,但从长远和发展的角度来看,使用QbD的企业可以走得更远。
参考资料
1.ICH Q6A的中英文版本
2.ICH Q8的中英文版本
3.制药质量的设计:产品和工艺开发,理解和控制
4.QbD被认为是一种国际商业机会渠道,受到FDA的积极推动。