名词解释:
GDUFA(非专利药品使用费法案),非专利药品企业支付法案
Pdu fa(处方药使用费法案),处方药企业支付法案
PAS(事先批准补充),需要事先批准的补充申请。
一、GDUFA产生背景
GDUFA于2012年10月1日由美国国会通过。在此之前,法律(即PDUFA法案)只要求NDA生产商在申报NDA时支付费用。然而,随着仿制药申请数量的增加,FDA有限的监管资源已经不能满足日益增长的仿制药申请需求。2012年,新仿制药申请积压量约2800件,而仿制药审评时间近30个月,远少于新药(新药审评时间:标准审评12个月,优先审评6个月)。
为了使公众更快获得安全有效的仿制药,降低成本,美国国会借鉴了实施PDUFA的成功经验,制定了GDUFA。
GDUFA要求制药公司向FDA支付仿制药申请的审查费和检查设施费用,帮助FDA增加资源和人员以减少积压,缩短仿制药申请安全性审查所需的平均时间,增加风险检查。相应的,FDA承诺加快仿制药的审评速度,增加审评过程的透明度和可预测性。
二、GDUFA的适用范围及费用类型
GDUFA要求所有在美国销售的制剂和原料药的生产商都必须支付一定的费用,该费用包括以下几个方面:
1.积压费用
2012年10月1日前向FDA提交申请但尚未获得批准的生产商需要提交一次性积压处理费,该费用仅适用于GDUFA实施的第一年(2012年10月1日-2013年9月30日)。
2.DMF费用
ANDA或PAS在2012年10月1日或之后首次引用的API DMF需要支付一次性费用,无论该DMF之前是否经过FDA的审核。
3.ANDA和考绩制度申请费
自2012年10月1日起,每份ANDA/pa在提交时都需要付费。
4.场地费
ANDA/ANDA/PAS涉及的制剂和原料药工厂需要自己注册并支付年费(宠物制造商除外)。
三、GDUFA的更新和收费标准
GDUFA每五年更新一次,但收费标准每财年更新一次(FDA财年为10月1日至次年9月30日)。
费用标准是根据年度通货膨胀率和CPI指数调整的总费用,除以当年ANDA/PAS申请的估计数量和工厂总数。
在FDA的内部评估中,会结合上一年的申请和注册工厂的数量。估算过程需要提交给国会,并通过联邦登记册向公众报告。下图显示了GDUFA费用计算方法的示例。
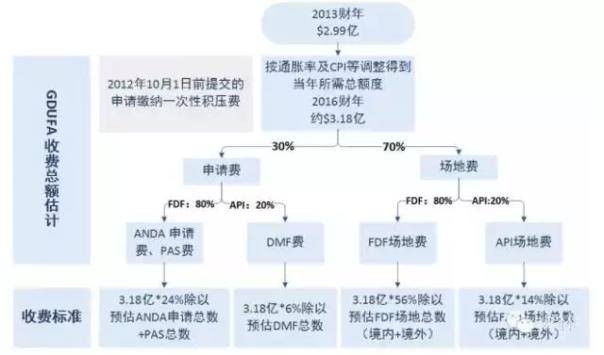
从上图可以看出,GDUFA收费总量是不变的。申请越少,注册工厂越少,每个申请或每个工厂的成本就越高。
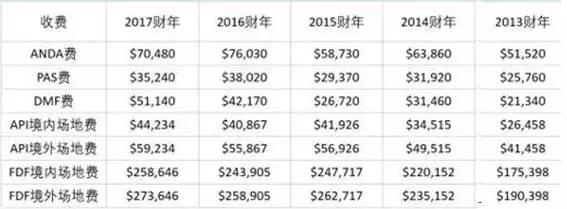
FDA的GDUFA费用表(2013-2017财年)
如图所示,2017年的费用比2016财年低,一个ANDA申请减少5000多美元。原因是FDA预计2017财年提交的ANDA数量将会增加。
四、GDUFA对行业带来的好处及影响
GUFDA是制药行业和FDA谈判的结果。谈判的结果是,行业同意支付上述费用,FDA做出了一系列承诺:提高评价效率、增加过程透明度、解决积压等一系列量化的绩效指标,具体如下:
1.指标1:解决积压问题。
FDA承诺在2017年底前完成90% ANDA申请的审查、批准前的变更、批准后的修订。FDA 2016年年报显示,FDA已经提前完成了这一目标。
2.指标2:提高效率
2.1提高ANDA审查的效率(anda审查效率增强)
2.1.1完整的回应函。
从2012年10月1日开始,FDA通过向企业发出完整的回函,及时与企业进行沟通。完整的回函包括FDA评估部门和不同评估专家对ANDA申请的完整意见。对于专家在ANDA评审过程中一些容易改变的缺陷,FDA会迅速通过电话与企业沟通,让企业及时纠正一些容易改变的缺陷。
2.1.2加强与企业的沟通(受控通信)
对FDA 2015年目标的承诺,即在收到ANDA申请后4个月内完成70%的信息沟通;2016年收到ANDA申请后2个月内完成70%的信息沟通;2017年收到ANDA申请后2个月内完成90%的信息沟通。
2.1.3电话会议(电话会议)
电话会议是FDA提供的另一种方式,方便仿制药厂商及时更正信息。在第一轮完整的回函发出后的10个工作日内,ANDA制造商可以通过书面申请与FDA进行电话会议,以解释或澄清回函中的相关问题。
2.1.4发布拒绝接收标准的指导方针。
此外,FDA还发布了拒绝接收标准,告知药企哪些缺陷会导致ANDA申请被拒绝(如果ANDA被拒绝,FDA将扣除25%的申请费,约10万人民币)。因此,为了避免资金的浪费,申请人需要确保ANDA申报不会被FDA拒绝。
2.1.5在专利质疑中加快对仿制药申请的审查(第IV段)
在GDUFA实施的前两年,FDA将加快审查有专利挑战的仿制药申请。详情请参考CEDER的MAPP 5240.3。
2.1.6 ANDA评估指标
GDUFA每五年更新一次。考虑到前期申请积压,GDUFA实施的前两年主要是解决文件积压的问题,尤其是加快挑战专利的仿制药申请。自GDUFA实施的第三年起,FDA承诺在收到ANDA申请后的15个月内对60%的申请采取行动。从第四年起15个月内对70%的申请采取行动;在第五年的10个月内对ANDA 90%的地区采取行动。评估期间,申请人提交变更或修改申请的,评估时间根据变更性质另行计算(限于篇幅,此处不再赘述)。不管怎么说,从最初ANDA审查时间的30个月甚至更长到10个月,这确实是一个很大的进步。
2.2提高DMF审查的效率(DMF审查效率增强)
FDA将采取与ANDA申请审查相同的方法加快DMF的审查,包括进行完整性评估,早期通过电话沟通DMF文件中容易改变的缺陷,并通过电话会议在DMF审查的回应函中解释相关问题。
3.指标3:现场检查(检查标准)
FDA承诺结合基于风险评估的科学方法,每两年对国内外药厂或原料药厂进行现场cGMP检查。也就是说,检查的频率可以根据风险评估的结果进行调整。首先将检查三种类型的工厂:
高风险工厂首先安排检查;
从未被FDA检查过的工厂也会优先检查;
提交了ANDA和ANDA文件的工厂已经过审查,可以批准或临时批准。
检查的最终目的是平等对待美国境内和境外的工厂,不放松对境外工厂的监管。因此,从现在开始,DMF或ANDA制造商应该注意它。在日常工作中加强cGMP培训时,他们应该始终牢记数据完整性的重要性。该写的SOP要写,而且要及时写。不确定那天FDA会不会来检查。当然,对自己的产品负责更重要。
4.指标4:促进监管科学的发展。
创造更多评估产品安全性、有效性、质量和性能的新工具、标准和方法。继续开发口服吸入、皮肤疾病局部应用和胃肠道药物产品的生物等效性新方法,继续开发基于科学的产品开发和上市后仿制药产品评价方法。例如,2016年发布了22个新的特定产品生物等效性指南,修订了19个指南。目前有15个吸入剂产品指南供药企参考。
五、GUFDA缴费的时间及不缴费的后果
如果申请人未能及时付款会怎么样?GUFDA制定了以下规定:
1.如果在提交ANDA后20个日历日内未缴纳ANDA费用,则视为申请未被接受。
2.如果ANDA报价的DMF没有及时支付,ANDA将被拒绝。
3.如果ANDA的生产站点出现故障